RADseq_Containers
User-guide for users of the RADseq Containerized Pipelines (HCMR)
Restriction-site associated DNA sequencing is a fractional genome sequencing strategy, that samples at reduced complexity across target genomes. Through the use of a restriction nuclease and attaching a series of adapters to the resulting DNA fragments, large numbers of genetic variations such as SNPs can be readily identified from analysis of next generation DNA sequence data. One can use a single or double-digest restriction site-associated DNA sequencing using the according protocol. The following workflows refer to ddRAD analyses.

What you will find and what will you need to use it
The pipelines are made using Snakemake and containerized through Singularity with the help of the Conda package manager. If you are not familiar with any of these tools, don’t worry. All you have to know, is that these technologies, give you the opportunity to transfer the workflow on any machine, as long as it works with a linux-based kernel and has Singularity installed. If these apply, you can run the pipeline effortlessly, without worrying about releases and packages that may not be compatible with each other. Conda handles that, and creates environments for the Snakemake workflow to run, without interferring with the system you are hosting it into.
The marriaging of these three tools, create the pipelines and deliver them to you in the form of container images. Each ready to use pipeline is represented by an image, and can be run in any cluster or environment that supports the resources and 3d parties tools needed.
In order for you to have some impact on the analysis and to also include your preferences in the procedure, the including and careful filling of a configuration file with lots of parameters is mandatory.
When it comes to the analysis itself, you can choose between a de-novo or a reference based approach, who’s steps is clesrly defined in the STACKS tool, the main algorithm used in such analyses. The following diagram summarizes the steps and inputs of each choice:
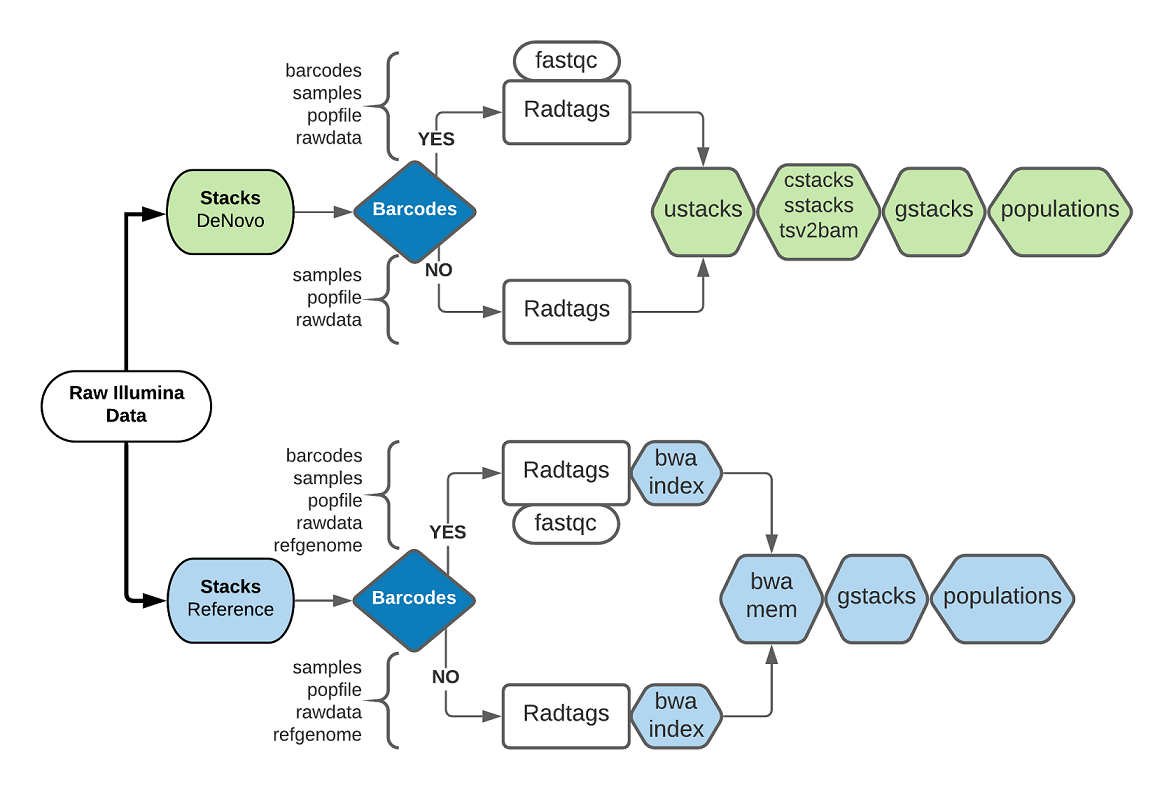
A. ddRAD_DN
This image is suitable for those who want to run a De Novo analysis with their reads. It performs a quality check on the raw reads, and then preprocessing through radtags, to proceed to ustacks and the rest of the STACKS pipeline. The pipeline can either perform demultiplexing or accept demultiplexed data.
The results of the image are the following:
Raw Data
- Quality reads for each raw read (R1,R2), generated by fastqc and located in the raw data directory.
- One directory that contains the results of the preprocessing of the raw short reads, named process_out.
- One directory that contains the results of the rest of the pipeline, named stacks_out.
- One directory located inside stacks_out, that contains the results of the final Populations step, named accordingly. Contains a folder for each combination of parameters given.
Demultiplexed Data
- Files that contain the results of the quality radtags run on the demultiplexed reads, in the input process_out dir.
- One directory that contains the results of the rest of the pipeline, named stacks_out.
- One directory located inside stacks_out, that contains the results of the final Populations step, named accordingly. Contains a folder for each combination of parameters given.
Here are the tools used for this analysis:
| Tools | Description | Version |
|---|---|---|
| fastqc | Quality check (Runs only with Raw Data) | 0.11.8 |
| stacks | Analysis | 2.34 |
B. ddRAD_Ref
Ref_ddRAD is an image suitable for those who want to run an analysis using a refernce genome. It performs quality checking on the raw reads, preprocessing using radtags, mapping to a reference through BWA, and last the gstacks and populations steps of STACKS. The pipeline can either perform demultiplexing or accept demultiplexed data.
The results of the image are the following:
Raw Data
- Quality reads for each raw read (R1,R2), generated by fastqc and located in the raw data directory.
- One directory that contains the results of the preprocessing of the raw short reads with radtags, named process_out.
- One directory that contains the results of the rest of the pipeline, named stacks_out.
- One directory located inside stacks_out, that contains the results of the final Populations step, named accordingly. Contains a folder for each combination of parameters given.
- One directory that contains the results of the BWA algorithm, named Alignments.
Demultiplexed Data
- Files that contain the results of the quality radtags run on the demultiplexed reads, in the input process_out dir.
- One directory that contains the results of the rest of the pipeline, named stacks_out.
- One directory located inside stacks_out, that contains the results of the final Populations step, named accordingly. Contains a folder for each combination of parameters given.
- One directory that contains the results of the BWA algorithm, named Alignments.
Here are the tools used for this analysis:
| Tools | Description | Version |
|---|---|---|
| fastqc | Quality check | 0.11.8 |
| bwa | Indexing & Mapping (mem) | 1.7.4 |
| stacks | Analysis | 2.34 |
Thus, in order for you to run your analysis in your organism’s or industry’s server, all you need is:
- A server that has Singularity installed
- The image of the pipeline you wish to use for your analysis
- The corresponding configuration file along with the image, that lets you define different parameters for the analysis
- A script that submits the image as a job into the nodes of your cluster
Breaking down the steps that lead to the analysis
Having found the image that correspond to the analysis you need, download it and copy it along with the configuration file in the home folder of your cluster account. A simple cp or a scp will do the trick. Once you’re done, open the configuration file with nano to edit it. The configuration file is probably the most important component for your analysis to work, so take your time and double check all the paths and the parameters needed here. The file is made to work for all the currently available analyses, so your intervention and filling will in fact let the whole system know which image you are going to use and thus the goal of your chosen pipeline, so make sure to fill the right variables in order for your image to be executed properly. Inside the configuration file you will also find some requirements that your raw data should fulfill. If your data or data directories do not follow the standards, please make the appropriate changes before editing the configurations. The cleaness of your directories is also very important. The workflow generates many intermediate files while running, and deletes them when not using them anymore, in order for your outputs to be carefully and tidy arranged, and your account free of unnecessary files. In order for the workflow to not mistakenly read or delete a file with the same name or regular expression of the file it is truly looking for, the directory where you copy the image and the configuration file into should be empty, and the directories of your raw data should not contain anything more or less than the data itself and the absolutely necessary input files asked. Clean the necessary directories before filling the configuration file, to avoid any mistakes. If you wish to re-run the workflow for any reason (errors or verifications), make sure to delete or move any already created output before proceeding.
Once done with all that, it’s time to let the automated workflow do the rest for you. Follow the standars for running a job on your server’s cluster and submit the image as follows (replacewith the name of the actual image you have chosen, and add any path needed): singularity run <image.simg>When the workflow is done, check carefully if all the files that should have been spawned are present in your directories, as and their status in the Summary.txt file. Ignore any other file mentioned, that may be spawned intermediately and has already been deleted by the workflow a priori.
Pipelines:
If you wish to use our images, get in contact with us for the necessary files.
Developers:
If you are a developer and you wish to explore our code and learn more about these technologies, ask for access to our GitHub repository for developers and maintainers.
Credits and Citations:
- Köster, Johannes and Rahmann, Sven. “Snakemake - A scalable bioinformatics workflow engine”. Bioinformatics 2012.
- Kurtzer GM, Sochat V, Bauer MW (2017), Singularity: Scientific containers for mobility of compute. PLoS ONE 12(5): e0177459.
- Crist-Harif et al, Conda, GitHub repository: https://github.com/conda.
- Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079.
Please credit accordingly:
Author: Nellie Angelova, Bioinformatician, Hellenic Centre for Marine Research (HCMR)
Coordinator/ Supervisor: Tereza Manousaki, Researcher, Hellenic Centre for Marine Research (HCMR)
Contact: n.angelova@hcmr.gr