De-Novo Transcriptome Assembly Containerized Pipelines
Welcome to your ultimate guide for using ready to go, containerized workflows for analyzing transcriptome data.
So you have sampled your organism of interest, sequenced the data, and found yourself with a bunch of NGS reads that need to be analyzed. But, you don’t really know how to proceed, or, you do know, but you would rather spend your time writing your papers or examining other organisms in the meantime. In both cases, you are in the right place at the right time.
What you will find and what will you need to use it
The pipeline for transcriptome assembly analyses is made using Snakemake and containerized through Singularity with the help of the Conda package manager. If you are not familiar with any of these tools, don’t worry. All you have to know, is that these technologies, give you the opportunity to transfer the workflow on any machine, as long as it works with a linux-based kernel and has Singularity installed. If these apply, you can run the pipeline effortlessly, without worrying about releases and packages that may not be compatible with each other. Conda handles that, and creates environments for the Snakemake workflow to run, without interferring with the system you are hosting it into.
The marriaging of these three tools, create the pipelines and deliver them to you in the form of container images. Each ready to use pipeline is represented by an image, and can be run in any cluster or environment that supports the resources and 3d parties tools needed.
In order for you to have some impact on the analysis and to also include your preferences in the procedure, the including and careful filling of a configuration file with lots of parameters is mandatory.
When it comes to transcriptome analysis, you can choose out of three different images, depending on your needs. Here is a brief explanation for each one of them:
A. SQTQ
Short Quality-Trimming-Quality (SQTQ) is an image suitable for those who want to run a simple quality control for their short (Illumina) reads, before and after trimming them. It actually performs quality check on the raw data, trimming of the raw data and quality check all over again. The results of the image are the following, located in the same directory your raw data live in:
- One directory that contains the now trimmed short data, named trimmomatic_output
- One directory that contains the results of the quality control of the raw short reads, named Multiq_raw_report
- One directory that contains the results of the quality control of the trimmed short reads, named Multiq_trimmed_report
Here are the tools used for this analysis:
| Tools | Description | Version |
|---|---|---|
| fastqc | Quality check | 0.11.8 |
| Multiqc | Quality check | 1.6 |
| Trimmomatic | Trimming | 0.39 |
B. TransA
Transcriptome Assembly (TransA) performs assembling with the R1 and R2 trimmed reads, and after that, calculates some stats for the assembly and quality-checks it through BUSCO. The results of the image are the following, located in the same directory your raw data live in:
- One directory called Assembly, which contains:
- A directory named Trinity, which holds the final assembly called Trinity.fasta and several side files generated through during the process.
- A .txt file called Stats.txt with information about the assembly’s statistics.
- A directory called Busco_Results with the BUSCO’s spawned files and results during quality checking.
- A directory of the lineage downloaded and used in the BUSCO analysis, in both untared and tar forms.
Here are the tools used for this analysis:
| Tools | Description | Version |
|---|---|---|
| Trinity | Assembly | 2.8.5 |
| Samtools | Used for stats and analyzations. | 1.9 |
| Busco | Quality check | 3.0 (Internal Blast: v2.2) |
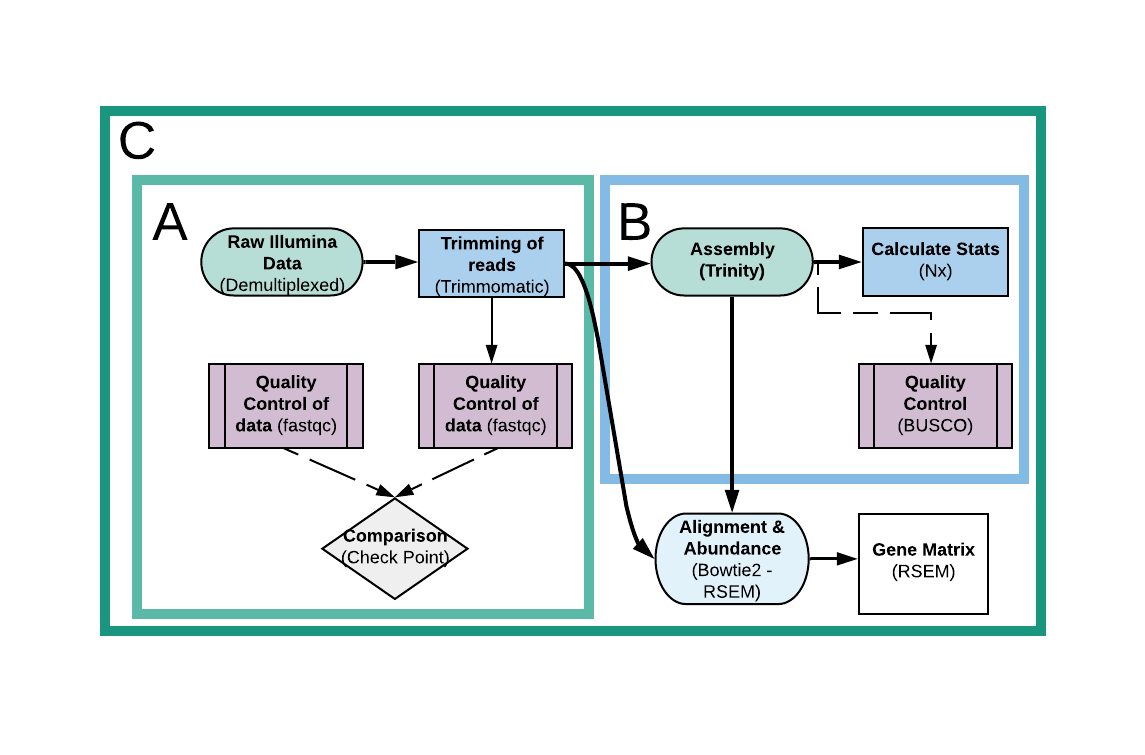
C. TransGM
Transcriptome Gene Matrix (TransGM) is an image suitable for larger analyses. It contains both SQTQ and TransA parts, and adds extra steps, which are an alignment and abundance estimation through Bowtie2 and RSEM, and a Gene Matrix construction through the latter, that can be later used for a downstream analyses as suitable (e.g. Differential Expression). Here are the main tools used by the pipeline, and all the results the image is spawning:
- One directory called Assembly, which contains:
- A directory named Trinity, which holds the final assembly called Trinity.fasta and several side files generated through Trinity during assembling and through Rsem and Bowtie2 in a later alignment and abundance estimation.
- A txt file called Stats.txt with informations about the assembly’s statistics.
- A directory called Busco_Results with the BUSCO’s spawned files and results during quality checking.
- A directory of the lineage downloaded and used for BUSCO analysis in both untared and tar forms.
- A directory called Rsem with the results of the alignment and abundance estimation for each sample.
- One directory called Gene_Matrix with the constructed gene matrix and some side files.
| Tools | Description | Version |
|---|---|---|
| Trinity | Assembly | 2.8.5 |
| Samtools | Used for stats and analyzations. | 1.9 |
| Busco | Quality check | 3.0 (Internal Blast: v2.2) |
| RSEM | Coupled with Trinity | 1.3.3 |
Thus, in order for you to run your analysis in your organism’s or industry’s server, all you need is:
- A server that has Singularity installed
- The image of the pipeline you wish to use for your analysis
- The corresponding configuration file along with the image, that lets you define different parameters for the analysis
- A script that submits the image as a job into the nodes of your cluster
Breaking down the steps that lead to the analysis
Having found the image that correspond to the analysis you need, download it and copy it along with the configuration file in the home folder of your cluster account. A simple cp or a scp will do the trick. Once you’re done, open the configuration file with nano to edit it. The configuration file is probably the most important component for your analysis to work, so take your time and double check all the paths and the parameters needed here. The file is made to work for all the currently available analyses, so your intervention and filling will in fact let the whole system know which image you are going to use and thus the goal of your chosen pipeline, so make sure to fill the right variables in order for your image to be executed properly. Inside the configuration file you will also find some requirements that your raw data should fulfill. If your data or data directories do not follow the standards, please make the appropriate changes before editing the configurations. The cleaness of your directories is also very important. The workflow generates many intermediate files while running, and deletes them when not using them anymore, in order for your outputs to be carefully and tidy arranged, and your account free of unnecessary files. In order for the workflow to not mistakenly read or delete a file with the same name or regular expression of the file it is truly looking for, the directory where you copy the image and the configuration file into should be empty, and the directories of your raw data should not contain anything more or less than the data itself. Clean the necessary directories before filling the configuration file, to avoid any mistakes. If you wish to re-run the workflow for any reason (errors or verifications), make sure to delete or move any already created output before proceeding.
Once done with all that, it’s time to let the automated workflow do the rest for you. Follow the standars for running a job on your server’s cluster and submit the image as follows (replace with the name of the actual image you have chosen, and add any path needed):singularity run <image.simg>When the workflow is done, check carefully if all the files that should have been spawned are present in your directories, as and their status in the Summary.txt file. Ignore any other file mentioned, that may be spawned intermediately and has already been deleted by the workflow a priori.
Note: The pipelines spawn a lot of files and data. Bare in mind that you should have enough space before running them in your repositories. The needed space varies, and depends on things such as the amount and size of your initial data.
Pipelines:
If you wish to use our images, get in contact with us for the necessary files.
Developers:
If you are a developer and you wish to explore our code and learn more about these technologies, ask for access to our GitHub repository for developers and maintainers.
Credits and Citations:
- Köster, Johannes and Rahmann, Sven. “Snakemake - A scalable bioinformatics workflow engine”. Bioinformatics 2012.
- Kurtzer GM, Sochat V, Bauer MW (2017), Singularity: Scientific containers for mobility of compute. PLoS ONE 12(5): e0177459.
- Crist-Harif et al, Conda, GitHub repository: https://github.com/conda.
- Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics, btu170.
- Philip Ewels, Måns Magnusson, Sverker Lundin, Max Käller, MultiQC: summarize analysis results for multiple tools and samples in a single report, Bioinformatics, Volume 32, Issue 19, 1 October 2016, Pages 3047–3048.
- Grabherr, Manfred G et al. “Full-length transcriptome assembly from RNA-Seq data without a reference genome.” Nature biotechnology vol. 29,7 644-52. 15 May. 2011, doi:10.1038/nbt.1883
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079.
- Felipe A. Simão, Robert M. Waterhouse, Panagiotis Ioannidis, Evgenia V. Kriventseva, Evgeny M. Zdobnov, BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs, Bioinformatics, Volume 31, Issue 19, 1 October 2015, Pages 3210–3212.
- Li, B., Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). https://doi.org/10.1186/1471-2105-12-323
Please credit accordingly:
Author: Nellie Angelova, Bioinformatician, Hellenic Centre for Marine Research (HCMR)
Coordinator/ Supervisor: Tereza Manousaki, Researcher, Hellenic Centre for Marine Research (HCMR)
Contact: n.angelova@hcmr.gr